Introductions
- Physiologic neonatal jaundice = Hyperbilirubinemia due to physiologic destruction of RBCs in the polycythemic newborn.
- Pathological neonatal jaundice = Cholestatic jaundice that persists beyond 2 weeks of age. These are conditions that lead to further investigations.
While up to 15% of breast-fed infants may experience prolonged jaundice lasting over 3 weeks, only a small fraction, approximately 0.04–0.2%, develop cholestatic jaundice. However, it is crucial to promptly investigate the specific etiology, as some conditions are associated with a poor prognosis. Among the causes of pathological jaundice in infants, biliary atresia warrants consideration due to its progressive and fatal nature. Early diagnosis, ideally before 2 months of age, is paramount as it can significantly improve the success rate of early surgical intervention (Kasai procedure). Conversely, conditions like neonatal hepatitis are often self-limited and typically do not necessitate surgical intervention.
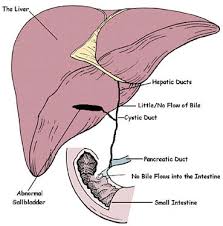
Biliary atresia (BA)
Patients usually present with signs of cholestatic jaundice including progressive jaundice, pale stools, hepatic enlargement, and dark urine within 2-6 weeks of life.
This condition arises from progressive fibrosis and obliteration of both intrahepatic and extrahepatic bile ducts. Without surgical intervention, it inevitably progresses to biliary cirrhosis. Patients typically succumb within the first years of life due to complications of biliary cirrhosis and end-stage liver disease.
Epidemiology
The prevalence of biliary atresia ranges from 1 in 5,000 to 20,000 live births. It is more frequent in East Asia compared to Western Europe. Gender predilection remains conflicting in recent literature. Full-term infants are more affected than premature infants.
Pathogenesis
The pathogenesis of biliary atresia is still unknown but is likely multifactorial, involving genetic, infectious (such as perinatal reovirus/rotavirus infection), and inflammatory factors that contribute to the development of obliterative cholangiopathy.
Classification
There are 2 forms of biliary atresia
- Non-syndromic form (80%)
- Syndromic form (biliary atresia splenic malformation syndrome (BASM); 20%)
Associated with- Polysplenia (or asplenia) ← very specific sign but low sensitivity
- Intestinal malrotation
- Preduodenal portal vein
- Interrupted inferior vena cava
- Aberrant hepatic artery
- Abdominal heterotaxia
- Congenital heart disease (such as ASD, VSD, and LV hypoplasia)
- Trisomy 18
Types
- Complete atresia of the extra-hepatic bile ducts (a)
This is the most frequent type (2/3 of patients). - Patent gallbladder with atretic cystic duct and extra-hepatic bile duct (b)
- Patent gallbladder, cystic duct and choledochus with atretic main common bile duct (c)
- Cystic forms with macrocyst at the liver hilum and variable atresia of the gallbladder and the extra-hepatic bile duct (d-g)

In all different subtypes of biliary atresia, intrahepatic bile duct fibrosis is always present. This explains the absence of bile duct dilatation despite impaired bile duct flow.
Investigations
Laboratory
- ↑ gamma-glutamyl transpeptidase (GGT)
- ↑ matrix metalloproteinase-7 (MMP-7)
Histology
- Portal or perilobular fibrosis
- Small intrahepatic bile ducts proliferation
- Absence of multi-nucleated giant cells in liver
- Bile plugs
- Edema
- Preservation of the basic hepatic lobular architecture
These findings are non-specific and can be found in many type of cholestasis including:
- Neonatal hepatitis
- Idiopathic (self-limiting)
- Infection: Hepatitis A/B, CMV, rubella, toxoplasmosis
- alpha-1-antitrypsin deficiency
- Familial recurrent cholestasis
- Other metabolic disorders
- Bile-plug syndrome
- Sepsis
- Hemolytic disorders
- Parenteral-nutrition-associated cholestasis
- Cystic fibrosis
- Alagille syndrome
- Choledochal cyst
Given the overlap of clinical, laboratory, and histopathological findings with other causes of pathological neonatal jaundice, imaging plays a crucial role in early diagnosis, leading to early intervention and improved patient outcomes.
Proposed algorithm for biliary atresia evaluation
According to the European Society of Paediatric Radiology (ESPR) Abdominal Taskforce at the 2019 meeting in Helsinki, the flowchart is as follows:

This flowchart is in accordance with the ESPGHAN and NAPGHAN guidelines which states;
- Postnatal US is useful to exclude other causes of cholestasis jaundice such as choledochal cyst, intrahepatic bile duct dilatation and gallstones.
- Postnatal US is useful to demonstrate features suggestive but not diagnostic of biliary atresia and a normal US does not rule out biliary atresia.
Ultrasonography findings
More specific signs
Gallbladder ghost triad
- Small (< 15-19 mm in length)
- Irregular shape
- Gallbladder length to width ratio of > 5.2
- Discontinuous echogenic wall
Absent of extra-hepatic bile ducts
Triangular cord sign
Fibrotic remnant of hepatic duct anterior to anterior right portal vein near porta hepatis.
- Cutoff thickness 3 – 4 mm
Main hepatic artery diameter > 2 mm
Specific ultrasonographic findings of biliary atresia
- Gallbladder ghost signs
- Absent of extra-hepatic bile ducts
- Triangular cord sign (3-4 mm)
- Main hepatic artery diameter > 2mm
Less specific signs
Subcapsular flow
Telangiectatic capsular/subcapsular small vessels
Other imaging techniques
Hepatobiliary scan
Radiopharmaceutical: Tc-99m DISIDA
Excretion to the intestine effectively excludes biliary atresia. Conversely, the absence of excretion can be attributed to any extrahepatic occlusion. (High sensitivity, low specificity)
Limitation
A pretreatment period of 5 days with phenobarbital is required for optimal accuracy. This may not be preferred for time-sensitive diagnosis of biliary atresia.
Shear wave elastography
Evaluate liver stiffness; shear wave velocity is significantly ↑ in biliary atresia. However, a normal elastography does not rule out biliary atresia.
MRCP
Key difference compare with ultrasonography
- Triangular cord cutoff > 5 mm
- Absent common bile duct seen in 25-40% of normal infants < 3 months old
ERCP
Problem solving tool in difficult cases but invasive and has significant morbidity and failure rates.
Intraoperative cholangiogram
The gold standard for diagnosing biliary atresia involves cannulation to the gallbladder and contrast injection. However, this procedure can only be done if there is a gallbladder remnant on previous ultrasonographic exam. In the absence of duodenal drainage, a liver biopsy is obtained with a presumptive diagnosis of biliary atresia. Subsequently, the patient may proceed to the Kasai operation.
Treatment
Kasai hepatoprotoenterostomy
- Resection of choledocal remnants, gallbladder, and portal plate
- Construction of a jejunal Roux-en-Y anastomosis
Success rate (achievement of bile flow)
- before 60 days of life → 70%
- after 90 days of life → 20%
This highlights the importance of rapid diagnosis before advanced cirrhosis develops, as advanced cirrhosis can contraindicate the Kasai surgery.
Liver transplantation
Indications:
- Fail Kasai operation
- Advanced cirrhosis
Most patients ultimately require liver transplantation. Biliary atresia is the most common reason for pediatric liver transplant.
Leave a Reply